Prion hay thể đạm độc là một loại protein bị gấp sai cấu trúc (misfolded), có khả năng truyền cấu trúc gấp sai này cho các biến thể bình thường cùng loại protein và kích hoạt tử vong tế bào.
Các Prion gây ra các bệnh prion được biết đến với tên gọi transmissible spongiform encephalopathy (TSE), là các bệnh về thoái hóa thần kinh (neurodegenerative) gây tử vong truyền nhiễm ở người và động vật.
Thể đạm độc còn thường gây sự biến đổi cấu trúc gấp sai một cách ngẫu nhiên, do đột biến gen, hoặc do tiếp xúc với một protein đã bị gấp sai.
1. Lịch sử nghiên cứu Prion
1.1. Ghi nhận đầu tiên
Từ “prion” bắt nguồn từ cụm từ “proteinaceous infectious particle” (hạt vi trùng protein), phản ánh khả năng tự truyền và lây nhiễm cấu trúc của nó sang các protein khác.
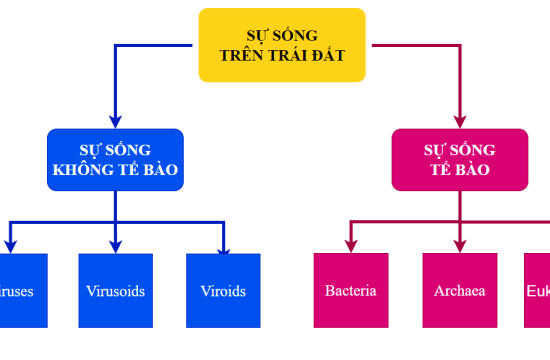
Điều này đặc biệt so với các tác nhân lây nhiễm khác như viroids, virus, vi khuẩn, nấm và ký sinh trùng, tất cả đều chứa acid nucleic (ADN, ARN hoặc cả hai).
Nguồn gốc nghiên cứu về “prion” bắt đầu từ những quan sát về bệnh scrapie, một loại bệnh thần kinh truyền nhiễm, được ghi nhận chủ yếu ở cừu và dê.
Trong thế kỷ 18 và 19, khi xem xét sự xuất khẩu cừu từ Tây Ban Nha, người ta phát hiện rằng bệnh scrapie thường xảy ra đồng thời.
Bệnh này làm cho động vật bị ảnh hưởng “nằm xuống, cắn chân và chân, cọ lưng vào cột, ngừng phát triển, ngừng ăn và cuối cùng trở nên khập khiễng”.
Dù nguyên nhân của bệnh chưa được biết rõ vào thời điểm đó, đây có thể là gọi là bệnh Prion hay transmissible spongiform encephalopathy (TSE), là bệnh gây tổn thương nặng đến hệ thần kinh ở động vật và con người đã lần đầu tiên được ghi nhận.

1.2. Những nghiên cứu sau này
Sau đó, trong thập kỷ 1950, Carleton Gajdusek tiến hành nghiên cứu và chứng minh rằng bệnh kuru, một bệnh truyền nhiễm đặc biệt, được xem là một dạng transmissible spongiform encephalopathy (TSE) có thể lây nhiễm cho tình dục thông qua một tác nhân gây nhiễm động vật có thể là một yếu tố lây nhiễm mới.
Carleton Gajdusek, một nhà nghiên cứu người Mỹ, đã giành giải Nobel y khoa năm 1976 cho công trình về bệnh kuru, một dạng transmissible spongiform encephalopathy (TSE).
Ông đã khám phá ra rằng bệnh kuru có thể được truyền từ người sang người, và điều này đã góp phần quan trọng vào hiểu biết về các bệnh prion.
Các nhà nghiên cứu London Tikvah Alper và John Stanley Griffith trong thập kỷ 1960 đã phát triển giả thuyết rằng transmissible spongiform encephalopathy (TSE) do một tác nhân gây nhiễm chỉ bao gồm các protein.
Họ đã đề xuất rằng một dạng bất thường của một protein tế bào có thể chuyển đổi các protein bình thường cùng loại thành dạng bất thường, dẫn đến quá trình nhân đôi.
1.3. Công trình đoạt giải nobel của Stanley B. Prusiner
Cuối cùng, vào năm 1982, Stanley B. Prusiner tại Đại học California, San Francisco, đã thông báo rằng nhóm nghiên cứu của ông đã tinh lọc được protein gây nhiễm giả định, không có mặt trong các máu khỏe mạnh.
Stanley B. Prusiner đã chứng minh rằng các bệnh transmissible spongiform encephalopathy (TSE) không phải do vi khuẩn, vi rút hoặc tác nhân truyền thống khác gây ra mà thực sự do một dạng đặc biệt của protein , được ông đặt tên là “prion” (proteinaceous infectious particle).
Ông đã cung cấp bằng chứng mạnh mẽ cho ý tưởng rằng prion có thể tự chuyển đổi và truyền nhiễm từ một protein thành một protein khác, gây ra các bệnh lý transmissible spongiform encephalopathy (TSE) như:
- Bệnh Creutzfeldt-Jakob (CJD) là một loại hiếm bệnh não gây ra sự hủy hoại nhanh chóng và không thể chữa trị ở con người
- “Bệnh đầu bò điên” (hay còn gọi là “mad cow disease”) là một tên gọi thông thường cho bệnh mất trí và suy giảm chức năng não ở bò, được gọi chính thức là “bệnh BSE” (Bovine Spongiform Encephalopathy).
Công trình của Prusiner đã khám phá cơ chế đột phá mà prion có thể gây ra bệnh lý và làm sáng tỏ một lỗ hổng lớn trong hiểu biết về bệnh lý truyền nhiễm. Công trình này đã thúc đẩy nghiên cứu sâu hơn về prion và đã có ảnh hưởng sâu rộng đối với lĩnh vực y tế và nghiên cứu về bệnh lý truyền nhiễm.
Protein này được đặt tên là “prion,” viết tắt của “proteinaceous infectious particle.” Phát hiện này đã đánh dấu bước tiến quan trọng và ông Prusiner đã giành giải Nobel Sinh lý học và Y khoa vào năm 1997 cho công trình về prion.
Từ đó, nghiên cứu về prion đã tiếp tục và đóng vai trò quan trọng trong lĩnh vực bệnh lây truyền và sinh học phân tử.
2. Gấp sai cấu trúc (misfolding) là gì?
Gấp sai cấu trúc (misfolding) là hiện tượng khi một protein không gập thành cấu trúc ba chiều bình thường mà thay vào đó gập thành một cấu trúc không đúng.
Cấu trúc ba chiều của một protein quyết định chức năng của nó. Khi protein gập sai cấu trúc, nó có thể mất đi khả năng thực hiện chức năng quan trọng và thậm chí có thể trở nên gây hại cho tế bào hoặc mô xung quanh.
Cấu trúc bình thường: Một protein bình thường có thể có một cấu trúc ba chiều đặc định, với các phần khác nhau được gập lại và nối với nhau một cách chính xác.
Gập sai cấu trúc: Trong trường hợp gập sai cấu trúc, protein có thể bị biến đổi thành một cấu trúc không đúng. Điều này thường xảy ra do các tác động ngoại lai như đột biến gen hoặc ảnh hưởng của các protein khác.
Trong trường hợp của prion, gập sai cấu trúc có thể được truyền từ protein prion gập sai tới các protein prion khác, dẫn đến sự tích tụ và tạo thành amyloid, gây tổn thương mô và tăng nguy cơ bị các bệnh prion. Điều này làm cho gập sai cấu trúc của prion có khả năng lây nhiễm và gây bệnh.
3. Protein prion (PrP)
3.1. Cellular Prion Protein (PrPC) và Scrapie Prion Protein (PrPSc)
Protein prion (PrP) là một loại protein có khả năng chuyển đổi từ dạng bình thường (PrPC) thành dạng bất thường (PrPSc), gây ra các bệnh transmissible spongiform encephalopathy (TSEs) hoặc bệnh prion.
PrPSc có khả năng lây lan và gây nhiễm nhanh chóng trong cơ thể.
Các thức phân loại của protein prion bao gồm:
PrPC (Cellular Prion Protein): Đây là dạng bình thường của prion protein, tồn tại ở khắp cơ thể người và động vật. PrPC có cấu trúc chủ yếu là α-helix và ít β-sheet. Nó không gây bệnh.
PrPSc (Scrapie Prion Protein): Đây là dạng bất thường của prion protein, chính là tác nhân gây bệnh prion. PrPSc có cấu trúc phức tạp hơn, với nhiều β-sheet và ít α-helix. PrPSc có khả năng lây nhiễm và làm thay đổi cấu trúc của PrPC khi chuyển đổi.
Cả hai dạng PrPC và PrPSc đều có cùng thành phần peptid, nhưng cấu trúc và tính chất của chúng khác biệt.
PrPSc có khả năng chống lại enzym protease và khó bị phân giải, đặc điểm quan trọng của protein gây bệnh prion. Sự chuyển đổi từ PrPC thành PrPSc là quá trình chính gây ra TSEs.
PrPSc có tỷ lệ cao hơn của β-sheet so với α-helix, và dạng bất thường này có khả năng tổ chức thành các sợi amyloid có cấu trúc cao hơn, gây ra sự kết tụ và tạo ra các mảng (plaques). Đây là những đặc điểm quan trọng của PrPSc, tác nhân chính gây ra các bệnh prion.
3.2. Protease-resistant PrPSc-like protein (PrPres)
PrPres (Protease-resistant PrPSc-like protein) là một dạng protein của PrPc (prion protein) được tạo ra trong điều kiện ngoài tổng sinh học, được biến đổi cấu trúc và chuyển đổi thành dạng kháng proteinase K. Proteinase K là một loại enzym có khả năng phân hủy các liên kết peptide trong các protein.
Dạng PrPres này khó bị phá hủy bởi enzym proteinase K, do đó được gọi là “protease-resistant” (kháng enzym phân hủy protein). Điều này phân biệt nó với PrPSc, một dạng khác của prion protein cũng chịu sự biến đổi cấu trúc, nhưng lại dễ bị phân hủy bởi proteinase K.
Các nghiên cứu về PrPres được thực hiện để nắm bắt hiểu rõ hơn về cơ chế gây bệnh và đặc tính của prion, đặc biệt trong bối cảnh bệnh transmissible spongiform encephalopathy (TSE). PrPres cũng được sử dụng để mô hình hóa quá trình chuyển đổi từ PrPC thành PrPSc, giúp hiểu rõ hơn về quá trình lây nhiễm và cách mà prion gây ra bệnh.
4. Bệnh Prion
Prion là một dạng đặc biệt của protein có khả năng gây ra nhiều bệnh với tên gọi transmissible spongiform encephalopathy (TSE) hay bệnh về thoái hóa thần kinh (neurodegenerative) ở người, động vật và gọi chung là bệnh prion.
4.1. Nguyên nhân gây ra bệnh
Nguyên nhân và cơ chế mà prion gây ra bệnh vẫn chưa được hiểu rõ đầy đủ, nhưng một điều chắc chắn là prion có khả năng tự chuyển đổi từ dạng protein bình thường sang dạng bất thường và lây lan trong cơ thể.
Chuyển đổi protein: Prion bình thường và prion bất thường là cùng một protein, nhưng ở dạng khác nhau.
Prion bình thường (PrP^C) có cấu trúc alpha-helix, trong khi prion bất thường (PrP^Sc) có cấu trúc beta-sheet.
Khi prion bình thường tiếp xúc với prion bất thường, nó có thể chuyển đổi thành prion bất thường.
Lây lan: Prion bất thường có khả năng lây lan bằng cách gây nhiễm trùng và thúc đẩy chuyển đổi của các phân tử prion bình thường thành prion bất thường.
Quá trình này tiếp tục lan rộng trong hệ thống thần kinh và làm suy yếu các tế bào thần kinh.
4.2. Một số bệnh do prion gây ra
Bệnh Creutzfeldt-Jakob (Creutzfeldt-Jakob Disease – CJD): Bệnh này ảnh hưởng đến hệ thống thần kinh, gây ra sự suy giảm chức năng não và các triệu chứng tâm thần.
Sốt Nhiệt đới Familiar Fatal Insomnia (FFI): Đây là bệnh hiếm gây ra sự mất ngủ và các triệu chứng tâm thần.
Bệnh Gerstmann-Sträussler-Scheinker (GSS): Gây ra suy yếu chức năng tâm thần và vấn đề về cảm giác.
Kuru: Loại bệnh prion đã được quan sát đầu tiên ở dân tộc Fore ở Papua New Guinea, thường được truyền qua việc ăn thịt người bị nhiễm prion.
Sởi điên (Scrapie): Ảnh hưởng đến cừu và dê, gây suy yếu và khó khăn vận động.
Các bệnh prion là những bệnh hiếm và đặc biệt nguy hiểm, không có phương pháp điều trị đặc hiệu cho đến nay. Hiểu thêm về cơ chế và nguyên nhân gây ra bệnh prion là mục tiêu quan trọng của nghiên cứu y học.
5. Cơ chế nhân bản của Prion
Sự lây lan hay nhân bản (replication) của prion đề cập đến quá trình mà prion, một loại protein không bình thường, tạo ra bản sao của chính nó thông qua chuyển đổi các protein bình thường thành dạng không bình thường, gây ra sự lây lan và gia tăng số lượng prion không bình thường.
Cơ chế nhân bản của prion được giải thích thông qua hai mô hình chính:
Heterodimer model:
- Mô hình này giả định rằng một phân tử PrPSc (prion không bình thường) kết hợp với một phân tử PrPC (prion bình thường) và thúc đẩy quá trình chuyển đổi PrPC thành PrPSc.
- Các phân tử PrPSc sau đó tách ra và tiếp tục chuyển đổi thêm phân tử PrPC, tạo ra sự lây lan và gia tăng số lượng prion không bình thường.
Fibril model:
- Mô hình này giả định rằng prion không bình thường (PrPSc) tồn tại chỉ dưới dạng sợi (fibrils).
- Đầu sợi fibril kết hợp với phân tử PrPC và chuyển đổi chúng thành prion không bình thường (PrPSc).
- Sự tăng mũi nhọn theo cấp số nhân của prion được giải thích bằng cách tính đến sự gia tăng kích thước và gãy sợi fibril.
Nhân bản của prion có ảnh hưởng lớn đến việc thiết kế dược phẩm. Do thời gian ủ bệnh của prion rất dài, một dược phẩm hiệu quả không cần loại bỏ toàn bộ prion mà chỉ cần làm chậm tốc độ tăng theo cấp số nhân.
Mô hình dự đoán rằng cách hiệu quả nhất để làm điều này là tìm ra một dược phẩm kết hợp với đầu sợi fibril và ngăn chúng mọc dài hơn.
Ngoài ra, các nghiên cứu đã chỉ ra rằng các phân tử đồng chủ (endogenous host cofactor molecules) như phospholipid và polyanions đóng vai trò quan trọng trong việc hình thành prion không bình thường có khả năng lây nhiễm cao trong vitro.